研究内容
遺伝子機能解析のツールとして広く使われるようになってきているゲノム編集で必要とされるデータ解析基盤技術の開発と、バイオインフォマティクス手法を駆使した遺伝子機能解析を行っています。 特に、データ駆動型ゲノム育種(デジタル育種)技術の開発に注力して研究をおこなっています。 また、生命科学分野のデータベース構築とその利用技術開発にも引き続き関わっています。
- 広島大学大学院統合生命科学研究科にて2023年3月24日に開かれた研究発表会でのポスター Genome Informatics Laboratory at Graduate School of Integrated Sciences for Life, Hiroshima University (March 2023) (DOI:
10.6084/m9.figshare.22298560)
その全容は、以下の図(bonohulab曼荼羅)のとおりです。
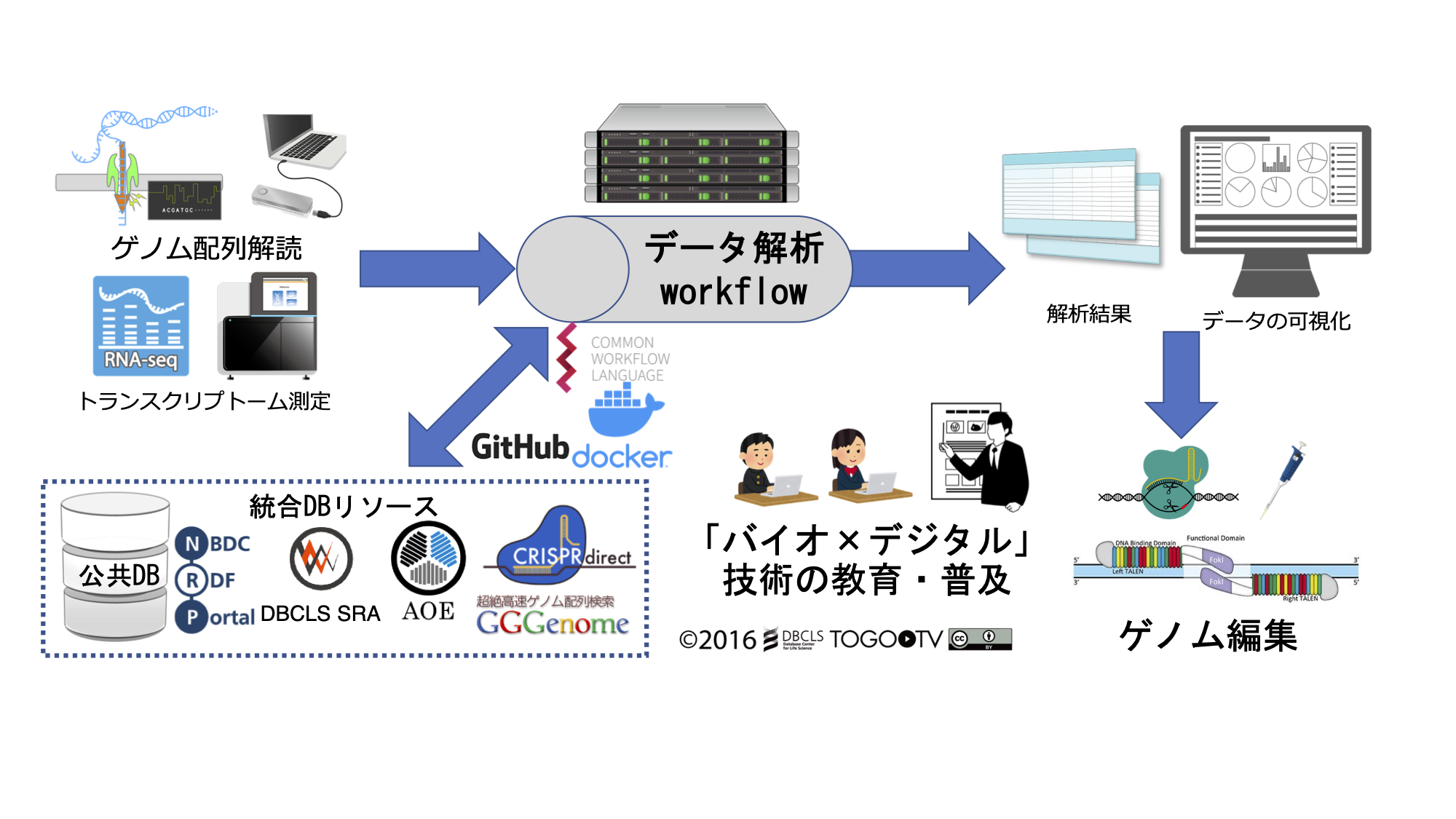
JST 共創の場形成支援プログラム(COI-NEXT)において広島大学が代表機関をつとめる「Bio-Digital Transformation(バイオDX)産学共創拠点」のもと、研究を進めており、以下のような研究が進行中です。 その他、民間企業との共同研究を複数並行して研究を進めております(主に 1.バイオDX基盤技術開発)。
学部学生や大学院生の研究テーマとしては、主に2. バイオインフォマティクスによる遺伝子機能解析を対象に取り組んでもらっております。
1. バイオDX基盤技術開発
データ駆動型ゲノム育種(デジタル育種)に必要なゲノム配列解読、トランスクリプトーム測定、ゲノム編集に持っていくためのデータ解析ワークフローなどの各種技術を開発します。 主にJST 共創の場形成支援プログラム 「Bio-Digital Transformation (バイオDX)産学共創拠点」(2022-2031年度)の支援に加えて、科研費 挑戦的研究(萌芽)「データ駆動型ゲノム育種を実現するデータ解析基盤技術の開発」(2021-2022年度)の支援のもと、進めております。
1.1. オミックスデータ測定・解析技術の開発
生物が持つ遺伝情報のすべて(ゲノム)、そこから発現した転写産物のすべて(トランスクリプトーム)、それらによって得られる代謝産物のすべて(メタボローム)の測定技術ならびにそのデータ解析手法を開発しています。
- 【広島大学プレスリリース】「トコジラミの殺虫剤感受性および抵抗性系統のゲノム解読〜抵抗性の発達に寄与する可能性のある遺伝子を発見〜」 (2024-10-18)
- 【広島大学プレスリリース】「赤シソの高精度なゲノム配列情報を決定 〜デジタル育種に向けた基盤情報を取得〜」 (2022/12/02)
1.2. ゲノム編集データ解析手法とその統合化ワークフローの開発
ゲノム編集のために必要な塩基配列解析手法を開発しています。
- 【広島大学プレスリリース】「ゲノム編集の成功を見きわめる!新ソフト『PtWAVE』で研究スピードをアップ」 (2025/05/27)
- 【広島大学プレスリリース】「AIがDNAを読み解いてミスを防ぐ! ゲノム編集で起こり得るRNAへの影響を予測する新システムを開発」 (2025/05/08)
- 【広島大学ウェブサイト】「ゲノム編集のための簡易的安全性評価ソフトウェアを新規開発〜バイオDXによる安全な品種改良やゲノム医療へ期待〜」(2023/09/25)
またゲノム編集による遺伝子機能解析を行う有用な物質を産生する昆虫に関して、アッセンブルしたトランスクリプトーム配列を入力として、ゲノム編集で利用する際に必要なデータ群を出力できるワークフローを構築します。 作成するワークフローは、公共データベースに登録されたゲノム編集関連データも含めてDBCLS/NBDCで開発しているRDFによるデータベース統合化技術を活用する他、Common Workflow Language (CWL)を用いて記述し、そのおかげでワークフローの再現性が保証されます。 ROIS-DS-JOINTの支援を受けた情報・システム研究機構 ライフサイエンス統合データベースセンターとの共同研究です。
- Oec N, Bono H (2021) Rapid metagenomic workflow using annotated 16S RNA dataset. DOI:
10.37044/osf.io/gbt8p - Bono H (2021) Meta-Analysis of Oxidative Transcriptomes in Insects. DOI:
10.3390/antiox10030345- SAQE: Systematic Analysis for Quantification of Everything
https://github.com/bonohu/SAQE
- SAQE: Systematic Analysis for Quantification of Everything
1.3. 有用物質生産パスウェイデザインシステムの開発
ゲノム・トランスクリプトーム測定データを使って、有用物質生産に関わる遺伝子群をアノテーション(注釈)するプログラムの組み合わせ(パイプライン)を構築します。 そして、それを利用してターゲットとなる遺伝子群をデジタルパスウェイ構築によって見出します。 現在は理化学研究所 生命医科学研究センター 生命医科学大容量データ技術研究チームと東京農工大学 大学院農学府 動物生化学(昆虫系)研究室との共同研究ですが、坊農の大学院時代からの研究テーマに源流のある研究です。
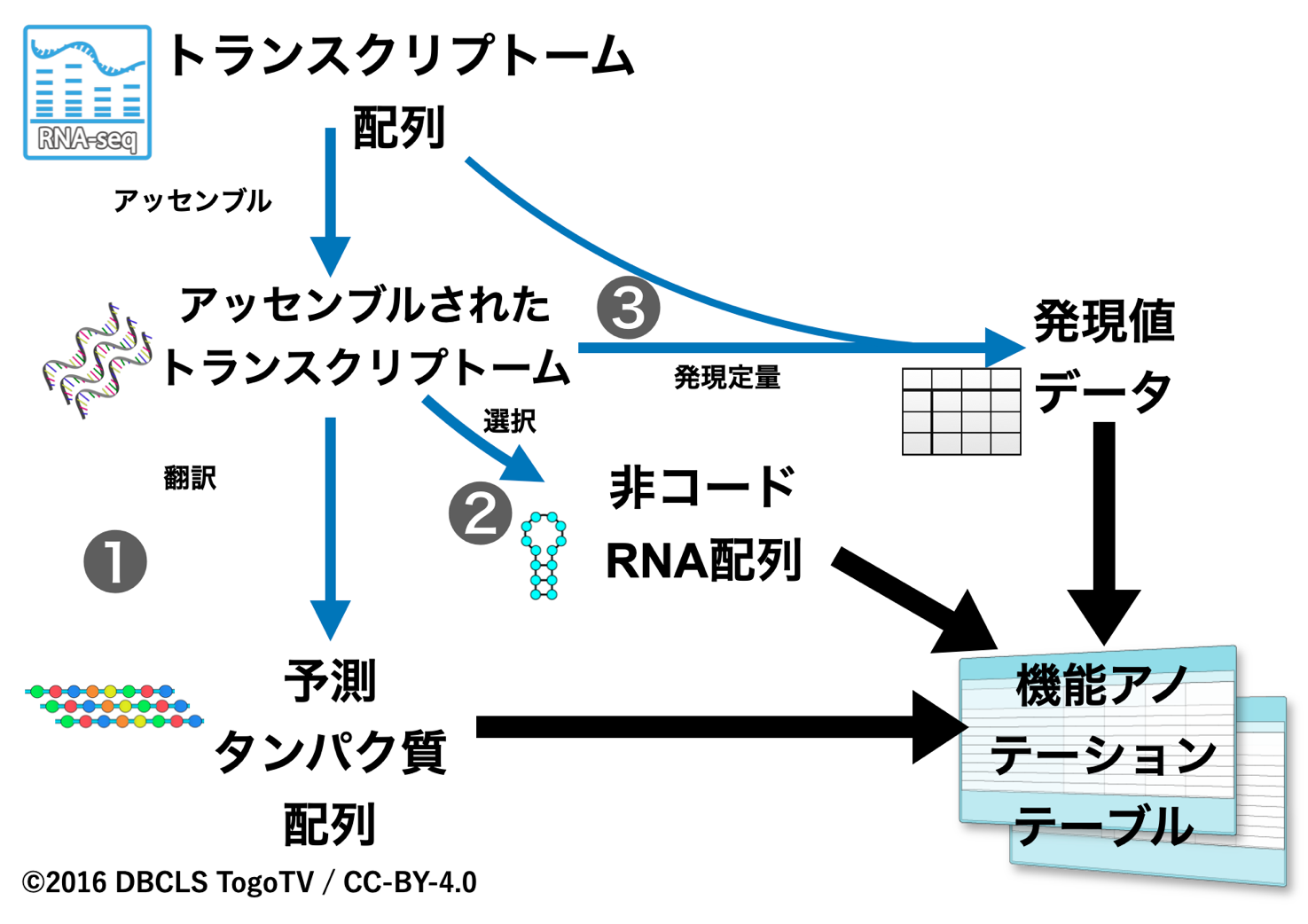
- 【広島大学プレスリリース「ゲノム編集のための昆虫遺伝子機能アノテーションワークフローを開発〜バイオDXによる昆虫機能利用に道~」】 (2022/07/07)
- Tabunoki H et al. (2013) Identification of key uric acid synthesis pathway in a unique mutant silkworm Bombyx mori model of Parkinson's disease. DOI:
10.1371/journal.pone.0069130 - Kasukawa T et al. (2003) Development and evaluation of an automated annotation pipeline and cDNA annotation system. DOI:
10.1101/gr.992803 - Bono H et al. (1998) Reconstruction of amino acid biosynthesis pathways from the complete genome sequence. DOI:
10.1101/gr.8.3.203
2. バイオインフォマティクスによる遺伝子機能解析
さまざまな生物種やデータに対してコンピュータを駆使したデータ解析手法を開発し、遺伝子機能にin sicilo (コンピュータ(シリコンチップ)の中で)に迫ります。 理学部生物科学科の学部生や統合生命科学研究科の大学院生は、主にこちらのテーマから取り組んでもらっています。
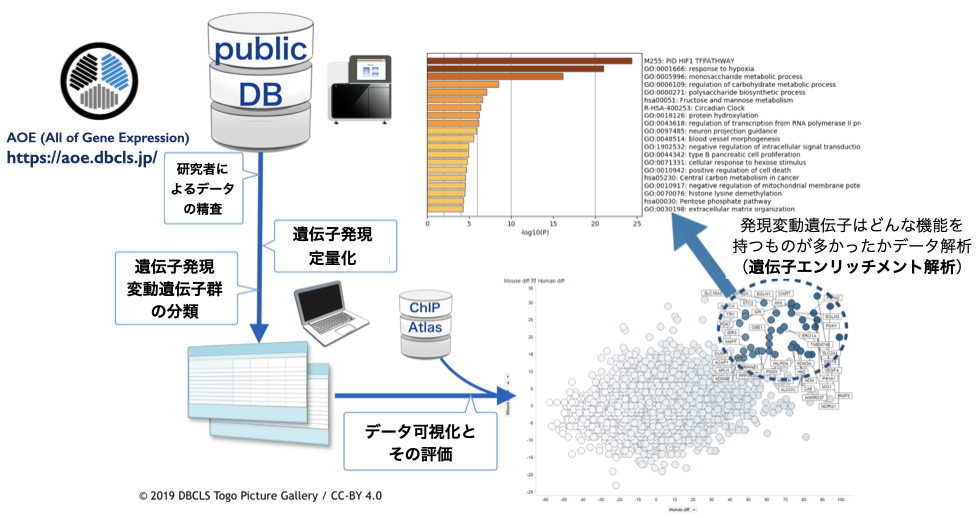
2.1. 公共データベースからのメタ解析
公共データベースに登録された複数のグループによる複数の研究の結果を統合し、より高い見地から分析するメタ解析を行っています。 特にストレス刺激の前後でのデータセットを作成してメタ解析することで新しい知見を得ようとしており、それらの成果をゲノム編集のターゲット情報として活用しようとしています。 これまで、低酸素刺激、酸化ストレス、熱ストレス、混み合いストレス、をターゲットとしてきました。
- 【広島大学プレスリリース】「イネなどの野生種の遺伝子に環境ストレス耐性のヒント!〜新品種の開発やゲノム編集への活用に期待〜」 (2025/08/01)
- 【広島大学プレスリリース】「公共データベースを活用したメタ解析により複数の家畜動物とそれらの原種間で発現が異なる遺伝子を複数同定〜データ駆動型ゲノム育種に向けた育種候補遺伝子の選出〜」 (2024/08/01)
- 【広島大学プレスリリース】「公共データベースを活用したメタ解析による植物におけるアブシジン酸関連ストレスの新たな知見〜大規模データの解析による環境ストレスに強い作物の開発に向けたゲノム編集ターゲット遺伝子を特定〜」 (2024/04/02)
- 【広島大学ウェブサイト】「公共データベースの利活用による高温に曝されると発生する熱ストレス応答の新たな知見〜気候変動に対応するためのデジタルトランスフォーメーション〜」 (2023/10/13)
- 【広島大学ウェブサイト】「社会性昆虫が繁殖を分業する仕組みに公共遺伝子発現データから迫る」 (2023/05/16)
- 【広島大学プレスリリース】「精密な転写産物参照配列セットを用いた低酸素応答性の評価-公共遺伝子発現データのデジタルトランスフォーメーション-」 (2022/10/13)
- 【広島大学プレスリリース】「昆虫の個体数密度に依存して姿・行動を変える能力に関係する遺伝子の同定」 (2022/10/07)
- 【広島大学プレスリリース】「冠水状態で植物の酸素取り込みが減少する「低酸素ストレス応答」のメカニズム解明につながる新たな候補遺伝子を発見」 (2022/07/28)
- 【広島大学プレスリリース】「メタ解析によって酸化ストレスと低酸素刺激の共通因子を同定」 (2021/12/07)
- 【広島大学プレスリリース】「マルチオミックス解析手法によって新規の低酸素応答性遺伝子を発見~公共遺伝子発現データと文献データのデジタルトランスフォーメーション~」 (2021/05/25)
その結果得られた発現変動遺伝子の情報をデータベース化したり、活用した共同研究を行っています。
2.2. デジタル育種に向けた比較ゲノム解析
デジタル育種に向けて、比較ゲノム解析に必要なデータ解析手法の開発を行っています。 これまでは農研機構 生物機能利用研究部門ほかとの共同研究で家畜昆虫であるカイコやミツバチ属(Apis)の比較ゲノム解析を行ってきました。
- 【広島大学プレスリリース】「カイコのシルク生産を担う絹糸腺における全遺伝子の発現量を解析、データを公開-カイコの産業利用拡大に貢献-」 (2025/05/30)
- 【広島大学プレスリリース】「公共データベースからのミツバチ参照遺伝子セットの構築〜農畜産生物のゲノム編集に向けたバイオデジタルトランスフォーメーション〜」 (2022/11/17)
- 【広島大学プレスリリース】「カイコ遺伝子発現データの拡張・公開~昆虫活用技術開発やデータ駆動型研究促進に期待~」 (2021/09/30)
また、九州大学 大学院医学研究院 応用幹細胞医科学部門 長寿幹細胞医学分野との共同研究においてがん化・老化耐性解明のための新しいモデル動物ハダカデバネズミのゲノムとトランスクリプトーム解析を行ってきました。
- 【JSTプレスリリース】「最長寿げっ歯類ハダカデバネズミでは老化細胞が細胞死を起こすことを発見~種特有のセロトニン代謝制御が鍵~」(2023/07/11)
- 【広島大学プレスリリース】「がん耐性齧歯類ハダカデバネズミの化学発がん物質への強い発がん耐性を証明~炎症抑制を介したがん耐性機構の一端を解明~」(2022/03/30)
3. 生命科学分野のデータベース構築とその利用技術開発
3.1. 公共データベース利用技術の開発
統合データベースプロジェクトで構築した遺伝子発現GEO目次をベースに、EBIのArrayExpressやDDBJのGenomic Expression Archive (GEA)のみならず、Sequence Read Archiveに納められた配列解読による発現データ(RNA-Seq)も検索できるようにした公共遺伝子発現データ目次(All of gne expression(AOE))を開発するなど、それらの基盤技術を活かした公共データベースの新しい利用技術の開発を行なっています。
- 【広島大学プレスリリース】「大規模言語モデル(LLM)をゲノム編集メタデータベース(GEM)に活用することで、情報が高精度に取得可能に!」 (2025/03/11)
- 【広島大学プレスリリース】「疾患における酸化ストレスに応答する新規候補遺伝子の探索手法を開発」 (2024/09/05)
3.2. FANTOM国際共同研究におけるデータベース開発
2000年から始まったFANTOMプロジェクト(当時は、Functional annotation of mouseの略で、現在はFunctional annotation of the mammalian genome)におけるデータベース開発とその活用技術開発を理化学研究所 生命医科学研究センター他との共同研究として行っております。
- Abugessaisa I et al. (2021) FANTOM enters 20th year: expansion of transcriptomic atlases and functional annotation of non-coding RNAs. DOI:
10.1093/nar/gkaa1054 - Bono H, Kasukawa T (2020) TogoEx: the integration of gene expression data. DOI:
10.37044/osf.io/esrc9 - Ramilowski JA et al. (2020) Functional Annotation of Human Long Non-Coding RNAs via Molecular Phenotyping. DOI:
10.1101/gr.254219.119 - Lizio M et al. (2015) Gateways to the FANTOM5 promoter level mammalian expression atlas. DOI:
10.1186/s13059-014-0560-6 - Carninci P et al. (2005) The transcriptional landscape of the mammalian genome. DOI:
10.1126/science.1112014 - Bono H et al. (2002) FANTOM DB: database of Functional Annotation of RIKEN Mouse cDNA Clones. DOI:
10.1093/nar/30.1.116 - Okazaki Y et al. (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. DOI:
10.1038/nature01266 - Kawai J et al. (2001) Functional annotation of a full-length mouse cDNA collection. DOI:
10.1038/35055500

Social